Antibody–drug conjugates (ADCs) are an emerging class of biotherapeutics that combine the specificity of monoclonal antibodies with the cytotoxic potency of small-molecule drugs. The antibodies are directed towards specific surface proteins on cancer cells, allowing precise delivery of the cytotoxic drug directly to tumor cells, thereby maximizing therapeutic efficacy while minimizing systemic toxicity to healthy tissues. A critical quality attribute (CQA) of ADCs is the drug-to-antibody ratio (DAR) distribution, which directly influences pharmacokinetics, stability and therapeutic efficacy. In cysteine-linked ADCs, conjugation occurs predominantly at inter-chain disulfide bonds, generating heterogeneous mixtures of antibody species bearing zero to four pairs of drug conjugates. Accurate characterization of this distribution is essential for process development, quality control, and understanding structure–function relationships.
Multiple Attribute Method (MAM) has emerged as a powerful LC–MS-based approach for the comprehensive characterization of monoclonal antibodies, enabling simultaneous monitoring of multiple CQAs at the peptide level such as post-translational modifications and glycosylations. However, for ADCs, this peptide-level approach could result in information loss such as the drug distribution profile. Therefore, a complementary approach based on intact mass analysis is necessary to provide both quantification of the different conjugated species and monitor the DAR.
In this study, we present a hybrid MAM LC–high-resolution mass spectrometry (LC–HRMS) workflow for the characterization of cysteine-linked ADCs, developed and optimized using a non-toxic antibody–drug conjugate mimic (“ADC-mimic”). In this ADC-mimic, the native cytotoxic payload is replaced with a dansyl-cadaverine–SMCC linker system, introducing a defined mass shift of 668 Da upon conjugation to cysteine residues. [LC1.1]The ADC-mimic was measured using reversed-phase chromatography on a Vanquish Flex UHPLC system coupled to an Orbitrap Exploris 240 mass spectrometer operating in an untargeted acquisition mode. Data was analyzed using the Thermo Scientific BioPharma Finder software.
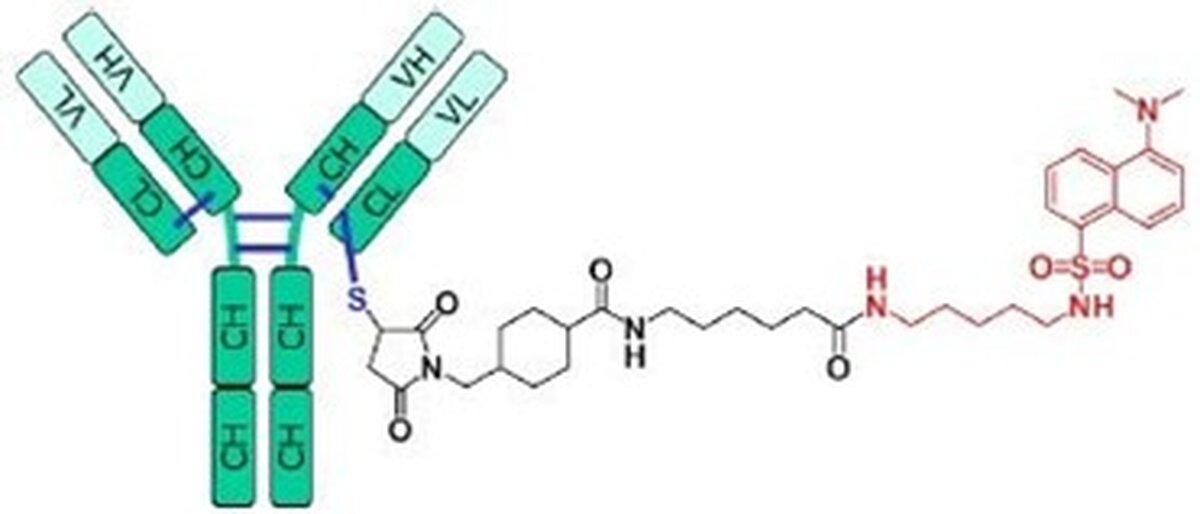
Before determination of the accurate masses of the ADC, the antibody was deglycosylated overnight using PNGase F treatment. Different conjugation states were defined after RP-C4 separation coupled to HRMS analysis. The DAR is calculated as the weighted sum of annotated peak intensities of the MS1 spectrum. For bottom-up analysis, the ADC mimic was reduced, alkylated, and digested with trypsin at 37 °C and peptides were analyzed using RP-C18 chromatography coupled to HRMS. After MS2 fragmentation, the precise amino acid containing the drug conjugate could be assigned. In addition, key PTMs relevant to ADC quality were monitored, including oxidation of methionine residues proximal to conjugation sites, deamidation of asparagine residues, aspartate isomerization, and N-terminal pyroglutamate formation. Relative abundances of glycoforms were also evaluated, allowing assessment of glycosylation heterogeneity.
This hybrid MAM approach provides a comprehensive characterization strategy, enabling simultaneous monitoring of site-specific modifications and accurate determination of DAR distribution.